
The nature of antigen recognition by TCR receptors
The adoptive transfer of cytotoxic T cells targeting intracellular tumor antigens has been intensively studied in recent years as a promising method of cancer immunotherapy. The ability of T cells to successfully eliminate threats by recognizing all kinds of foreign antigens depends on the enormous plasticity of their T-cell receptors (TCRs). What makes TCR different from chimeric antigen receptors (CAR) is that it recognizes a different type of antigen: protein fragments, which have been processed and displayed as short peptides (epitopes) on the surface of the antigen-presenting cell (APC), or target cell. Epitopes are presented by the major histocompatibility complex (MHC), which creates the immunological phenomenon known as MHC restriction. Discovered in 1974 by Zinkernagel and Doherty, their finding earned them the Nobel Prize in Physiology and Medicine in 1996. The major advantage of TCR-T cells over CAR-T cells is the ability to recognize a much broader spectrum of tumor antigens, not being restricted to extracellular proteins (Garber 2018) (Fig. 1). CAR-T cells are mostly tested and proven to be effective in the treatment of hematological malignancies, representing less than 10% of all types of cancer. Therein, finding a specific extracellular antigen like CD19 on the surface of B cells is easier than solid tumors. Another benet is that the toxic side effects connected with CAR-T cell therapies, like cytokine release syndrome or neurotoxicity are lower in TCR-T therapies (Xu 2018). However, there are reports of fatal off-target responses, as mentioned later in the article. The lower toxicity is related to the fact that (at least for some target types) TCRs preferentially recognize tumor antigens presented only by cancer cells; therefore (in an ideal scenario with no cross-reactivity), normal cells are not killed. In the case of CAR-T cells, for example, anti-CD19 CAR-T cells also destroy healthy cells, resulting in the elimination of healthy B cells (B cells aplasia).
An effective cytotoxic response is conditioned by a relatively high expression of the antigen on the surface of the tumor cells. Unfortunately, some cancers adopt escape mechanisms to avoid the immune response, including the impaired MHC class I expression (Hicklin 1999) or the loss of heterozygosity of MHC genes (Klippel 2014).
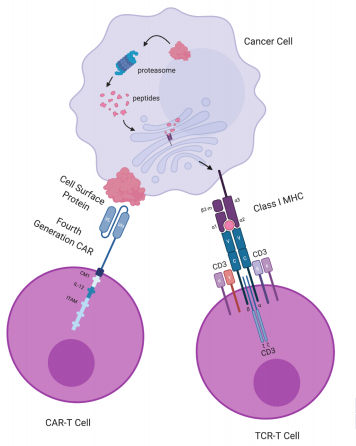
Figure 1. Target recognition diagrams for CAR-T-cell and TCR-T-cell therapies. CAR-T cells target cell surface proteins. CAR-T cells contain an scFv consisting of an antibody’s variable light (VL) and variable heavy (VH) chain, a transmembrane domain and a CD3ζ chain with ITAM for T-cell activation. The fourth-generation CAR (shown here) further includes a costimulatory molecule (CM1), such as CD28 and B7, and an interleukin-12 (IL-12) domain for enhanced T-cell effects. Receptor engineered T cells have a TCR consisting of an α chain and a β chain, each containing a variable domain (v) and a constant domain (c), as well as an unlabeled transmembrane domain and 6 CD3 chains for T-cell activation. A class I MHCprotein consists of an α chain with 3 domains: α1, α2, α3, and a β2-microglobulin. Class I MHC proteins present peptides derived from proteolysis of intracellular proteins. CAR: chimeric antigen receptor; ITAM: immunoreceptor tyrosine-based activation motif; MHC: major histocompatibility complex; scFv: single-chain fragment variant; TCR: T-cell receptor. Figure based on the article: The Emerging World of TCR-T Cell Trials Against Cancer: A SystematicReview used under CC BY 4.0 /
The graphic and colors have been changed from the original.
A brief historical overview of adoptive T cell therapies.
In the first article on our blog, titled “Future perspectives for CAR-T cell therapies”, we provided a brief historical introduction to anti-tumor T-cell therapies. As a reminder, the first clinical trials with T cells freshly isolated from cancer tissue samples of melanoma patients (also known as tumor-infiltrating lymphocytes, TILs) were conducted in the late 1980s by Rosenberg et al. (Rosenberg 1988). Complete and partial responses were observed in 11 of 20 patients with metastatic melanoma, and in approximately one-third of 86 patients of a larger, later study from 1994 (Rosenberg 1994).
Although the above-mentioned studies have undoubtedly demonstrated the possibility of using TILs in the treatment of cancer, no complete remission has been achieved and the median duration of response was only 4 months. This not fully satisfactory effect obtained by Rosenberg and his colleagues did not discourage them, instead, it only intensified studies of the adoptive T-cell therapies. Further research into the biology of cancer has shown that the immunosuppressive tumor microenvironment might suppress the immune response, resulting in ineffective tumor clearance. In addition, discoveries about processing, presentation, and recognition of tumor antigens have proven that affinity of the TCR for tumor antigen is key in recognizing a target cell (Stone 2009). Naturally occurring T cells with high affinity for tumor antigens are very rare due to the negative selection in thymus (Eisen 1996). Therefore, current research and clinical therapies focus on the use of gene-engineered T cells with affinity-enhanced TCRs. One method of obtaining such receptors is with the use of human peptide-major histocompatibility complex (HLA)-transgenic mice, where vaccination with a known tumor antigen generates high affinity, murine TCRs against such a target (Kuball 2005). A continuation of this approach was the development of transgenic mice expressing the entire human TCR repertoire by Blankenstein Lab (Li 2010). Currently, the T-knife biotech company conducts further research on TCRs for T-cell therapy of solid tumors and is approaching Phase I/IIa trials using autologous, gene-modified T-cells targeting the MAGE-A1 tumor antigen. Another way to generate a large number of high affinity receptors is by using the high throughput yeast and phage systems that rely on random mutagenesis (Holler 2000, Li 2005). Yet another approach that is used to obtain tumor-specific TCRs is the isolation of tumor-specific lymphocytes from cancer patients who, for example, had a good response to checkpoint inhibitor therapy and sequencing their TCRs. These TCRs subsequently can be cloned into an expression vector (e.g. retrovirus or the Sleeping Beauty transposon/transposase system). Autologous T cells isolated from the peripheral blood of the patient can be finally transduced with this expression vector, propagated in vitro to the desired quantity, and infused back into the patient (Bear 2012). All the techniques mentioned above have helped to mitigate some of the issues associated with the development of effective adoptive T-cell therapies, such as low affinity of endogenous TCRs and insufficient numbers of tumor-specic T cells.
Types of tumor antigens
Finding the right antigen is the holy grail of successful CAR-T and TCR-T therapies. With the broadened search space not limited to the cell’s surface, more and more targets for TCR-T therapies are being discovered.
There are many ways of categorizing epitope targets. For example, in to the Cancer Antigen Peptide Database, antigens encompassing multiple cancer types are grouped into five categories:
– mutation antigens: unique antigens which result from point mutations in genes that are expressed ubiquitously,
– tumor-specific antigens: shared antigens which are expressed in many tumors but not in normal tissues,
– differentiation antigens: shared antigens which are also expressed in the normal tissue from which the cancer originated,
– over-expressed antigens: shared antigens which are expressed in different healthy tissues and over-expressed in tumors,
– and the last group, potential antigens, which are candidates to the above-mentioned categories.
The targets from the category of mutational antigens (often called neoantigens) seem like an ideal target for tumor-specific TCR-T therapies that wouldn’t harm the healthy tissue. The problem is that often these neoantigens are unique and not shared among patients.
The off-target reaction
Some types of these antigens are safer in terms of being targeted in TCR-T therapies, while others, like differentiation antigens or over-expressed antigens, carry an increased risk of off-target side effects caused by T-cell cross-reactivity (Fig. 2).
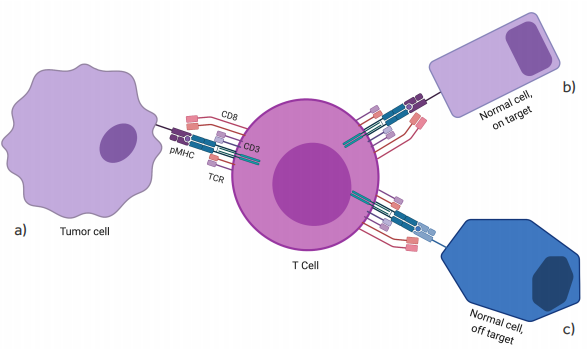
Figure 2.
On- and off-target recognition by TCR gene-modified T cells. Although TCR gene-modified T cells are designed to redirect antigen reactivity and maintain specificity, preclinical and clinical data have shown the potential for TCR-engineered T cells to exhibit (a) appropriate on-target, on-tumor antigen recognition (tumor eradication); (b) on-target, off-tumor recognition (i.e. low-level antigen on normal tissue); or (c) off-target cross-reactivity (i.e. related or unrelated antigen on target or non-target tissue)(Spear 2019).
Figure based on the figure from article: “Understanding TCR affinity, antigen specificity, and cross-reactivity to improve TCR gene-modified T cells for cancer immunotherapy”.
The graphic and colors have been changed from the original.
One of the first examples of off-target reaction was a clinical trial conducted in melanoma patients with the use of TCR-engineered, anti-MART-1 T cells with an enhanced affinity. Apart from an observed objective clinical response (~19%), high affinity T cells damaged also normal melanocytes in the skin, eye, and ear, which required local steroid administration to treat uveitis and hearing loss (Johnson 2009). Currently, over 100 clinical trials are being conducted with the use of TCR-T cells therapy. The results of the clinical trials targeting NY-ESO-1 in synovial sarcoma and myeloma patients seem to be very promising. One month after TCR transfer, 61% (11/18) of patients with synovial sarcoma had an objective clinical response, while 55% (11/20) of patients with metastatic melanoma experienced an objective clinical response (Robbins 2014).
The critical importance of carefully designing and testing an adoptive TCR-T therapy before entering clinical trials has been shown by a clinical trial where the first two treated patients developed cardiogenic shock and died within a few days of anti-MAGE-A3 affinity-enhanced T-cells infusion (Linette 2013). Interestingly, the reaction was directed against an unrelated protein – titin – which was off-target and found in striated muscles. The above-mentioned examples clearly show the dangers of off-target toxicity in these therapies and force researchers to develop methods to predict or experimentally probe the risk of the occurrence of off-target toxicities.
We hope that further research focused on improving TCR-T cell therapies will bring an effective and new solution to the fight against cancer.
- Zinkernagel RM, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974 Apr 19;248(5450):701-2. doi: 10.1038/248701a0. PMID: 4133807.
- Garber K. Driving T-cell immunotherapy to solid tumors. Nat Biotechnol. 2018 Mar 6;36(3):215-219. doi: 10.1038/nbt.4090. PMID: 29509745.
- Xu Y, Yang Z, Horan LH, Zhang P, Liu L, Zimdahl B, Green S, Lu J, Morales JF, Barrett DM, Grupp SA, Chan VW, Liu H, Liu C. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018 Nov 20;4:62. doi: 10.1038/s41421-018-0066-6. PMID: 30479831; PMCID: PMC6242878.
- Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today. 1999 Apr;5(4):178-86. doi: 10.1016/s1357-4310(99)01451-3. PMID: 10203751.
- Klippel ZK, Chou J, Towlerton AM, Voong LN, Robbins P, Bensinger WI, Warren EH. Immune escape from NY-ESO-1-specific T-cell therapy via loss of heterozygosity in the MHC. Gene Ther. 2014 Mar;21(3):337-42. doi: 10.1038/gt.2013.87. Epub 2014 Jan 23. PMID: 24451117; PMCID: PMC4040020.
- Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988 Dec 22;319(25):1676-80. doi: 10.1056/NEJM198812223192527. PMID: 3264384.
- Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994 Aug 3;86(15):1159-66. doi: 10.1093/jnci/86.15.1159. PMID: 8028037.
- Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. 2009 Feb;126(2):165-76. doi: 10.1111/j.1365-2567.2008.03015.x. PMID: 19125887; PMCID: PMC2632691.
- Eisen HN, Sykulev Y, Tsomides TJ. Antigen-specific T-cell receptors and their reactions with complexes formed by peptides with major histocompatibility complex proteins. Adv Protein Chem. 1996;49:1-56. doi: 10.1016/s0065-3233(08)60487-8. PMID: 8908296.
- Kuball J, Schmitz FW, Voss RH, Ferreira EA, Engel R, Guillaume P, Strand S, Romero P, Huber C, Sherman LA, Theobald M. Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity. 2005 Jan;22(1):117-29. doi: 10.1016/j.immuni.2004.12.005. PMID: 15664164.
- Li LP, Lampert JC, Chen X, Leitao C, Popović J, Müller W, Blankenstein T. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med. 2010 Sep;16(9):1029-34. doi: 10.1038/nm.2197. Epub 2010 Aug 8. PMID: 20693993.
- Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A. 2000 May 9;97(10):5387-92. doi: 10.1073/pnas.080078297. PMID: 10779548; PMCID: PMC25838.
- Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005 Mar;23(3):349-54. doi: 10.1038/nbt1070. Epub 2005 Feb 20. PMID: 15723046.
- Bear AS, Morgan RA, Cornetta K, June CH, Binder-Scholl G, Dudley ME, Feldman SA, Rosenberg SA, Shurtleff SA, Rooney CM, Heslop HE, Dotti G. Replication-competent retroviruses in gene-modified T cells used in clinical trials: is it time to revise the testing requirements? Mol Ther. 2012 Feb;20(2):246-9. doi: 10.1038/mt.2011.288. PMID: 22297819; PMCID: PMC3277217.
- Spear TT, Evavold BD, Baker BM, Nishimura MI. Understanding TCR affinity, antigen specificity, and cross-reactivity to improve TCR gene-modified T cells for cancer immunotherapy. Cancer Immunol Immunother. 2019 Nov;68(11):1881-1889. doi: 10.1007/s00262-019-02401-0. Epub 2019 Oct 8. PMID: 31595324.
- Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009 Jul 16;114(3):535-46. doi: 10.1182/blood-2009-03-211714. Epub 2009 May 18. PMID: 19451549; PMCID: PMC2929689
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013 Aug 8;122(6):863-71. doi: 10.1182/blood-2013-03-490565. Epub 2013 Jun 14. PMID: 23770775; PMCID: PMC3743463.
- Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Li YF, El-Gamil M, Rosenberg SA. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015 Mar 1;21(5):1019-27. doi: 10.1158/1078-0432.CCR-14-2708. Epub 2014 Dec 23. PMID: 25538264; PMCID: PMC4361810.



